Адреногенитальный синдром — механизм развития, симптомы и методы лечения
Данное заболевание относится к группе наследуемых нарушений синтеза стероидных гормонов. Распространенность адреногенитальных расстройств зависит от расовой и национальной принадлежности обследуемых. Среди европеоидов частота встречаемости составляет 0,007%, тогда как в некоторых семитских популяциях этот показатель достигает 17–19%.
Характеристика патологии
Заболевание поражает как женщин, так и мужчин, но в большинстве случае страдает от него прекрасный пол. У 90% пациенток с адреногенитальными отклонениями обнаруживается мутация гена, ответственного за выработку фермента 21-гидроксилазы и кортикостероидов.
Гормональный дисбаланс возникает еще во время внутриутробного развития плода и обнаруживается с момента рождения. Вследствие недостаточного синтеза кортизола у больных растет концентрация андрогенов. Основным клиническим проявлением адреногенитальных расстройств является выраженная вирилизация половых органов. У новорожденных девочек выявляются признаки женского псевдогермафродитизма.
При классических (врожденных) формах ферментопатии прогноз зависит от своевременности постановки диагноза и качества заместительной терапии. Длительно сохраняющаяся гиперандрогения повышает риск развития косметических дефектов, что нарушает психосоциальную адаптацию. В случае адекватного лечения женщин с классическими формами адреногенитальных отклонений возможно наступление беременности.

Механизм развития
В остальных случаях дефект ДНК приводит к нарушению производства других ферментов, участвующих в стероидогенезе – 3-β-гидроксистероиддегидрогеназы, 22-десмолазы, синтетазы альдостерона и др. Важно понимать, причины отклонений носят сугубо наследственный характер. Факторами риска синдрома принято считать:
- повышенный уровень ионизирующего излучения;
- прием сильнодействующих препаратов;
- острое либо хроническое радиационное отравление;
- интеркуррентные патологии;
- длительное использование гормональных контрацептивов;
- хирургическое вмешательство;
- дисфункция щитовидной железы;
- аборты;
- патологии почек;
- опухолевый процесс;
- травмы;
- стрессы.
В основе механизма развития классического (врожденного) варианта адреногенитальных нарушений лежит принцип обратной связи, начальным звеном которой является недостаток стероидов. Несостоятельность процесса гидроксилирования сопровождается неполным превращением прогестерона, а также 17-гидроксипрогестерона в дезоксикортикостерон и 11-дезоксикортизол. В результате уровень кортизола снижается. При этом усиливается выработка адренокортикотропного гормона (АКТГ), который, собственно, и вызывает компенсаторную гиперплазию коры надпочечников.
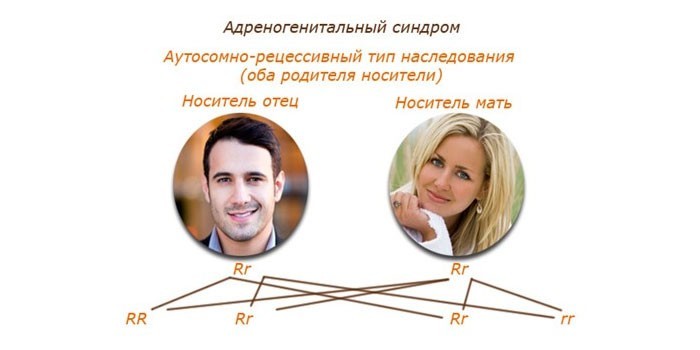
Риск наследования патологии
Вероятность рождения ребенка с адреногенитальным расстройством при носительстве патологического гена у обоих родителей составляет 25%, а в браке носителя и больного – 75%. При ситуации, когда один партнер имеет полноценное ДНК, клинические проявления синдрома у детей не обнаруживаются. Крайне редко адреногенитальные нарушения наследуются спорадически. Внезапное появление признаков синдрома, как правило, обусловлено нарушениями в процессе формирования женских и мужских репродуктивных клеток.
Особенности симптоматики
Сольтеряющая форма – самый тяжелый вариант течения АГС, который проявляется в первые годы жизни ребенка грубыми пороками развития половых органов у девочек и их увеличением у мальчиков. Дети с адреногенитальными расстройствами часто срыгивают, мочатся. Сольтеряющий синдром приводит к необратимым метаболическим нарушениям, обезвоживанию.
При классических формах расстройства степень вирилизации различается: от гирсутизма до яркого гетеросексуализма. Первые симптомы отклонений у младенцев женского пола обнаруживаются сразу после рождения. Вирильная форма у мальчиков проявляется позднее. Примечательно, что при рождении их половые органы правильно сформированы. Клинически синдром проявляется в 3–4 года. По мере взросления у мальчиков с гиперплазией надпочечников прогрессирует репродуктивная дисфункция.
Благоприятной в плане течения считаются неклассические формы адреногенитальных отклонений. Признаки скрытого синдрома проявляются во время полового созревания. При этом гениталии больных имеют вполне нормальное строение, расстройство выявляется случайно во время обследования по поводу бесплодия или нарушения менструального цикла. В зависимости от дефекта ферментных систем выделяют следующие виды адреногенитального синдрома:
|
Дефектный фермент |
Клиническая картина |
|
20, 22-десмолаза |
Нарушается секреция стероидов из холестерина в активные стероиды, что приводит к сольтеряющему синдрому, глюкокортикоидной недостаточности и летальному исходу. |
|
3-ол-дегидрогеназа |
Приводит к нарушению выработки кортизола и альдостерона, на фоне чего развивается синдром потери соли. За счет частичной выработки дегидроэпиандростерона вирилизация у девочек имеет слабовыраженный характер. |
|
17-гидроксилаза |
Проявляется дефицитом эстрогенов, андрогенов, кортизола. Провоцирует половое недоразвитие, гипертонию, гипокалиемический алкалоз. |
|
11-гидроксилаза |
Вызывает существенный избыток 1-дезоксикортикостерона. При этом варианте синдрома признаки вирилизации менее выражены. Отмечается высокое давление, задержка хлоридов, натрия. |
|
18-оксидаза |
Приводит к недостатку альдостерона. Клинически адреногенитальное расстройство проявляется сольтеряющим синдромом. Приводит к смерти в раннем детстве. |
|
21-гидроксилаза |
Тяжесть клиники синдрома обусловлена степенью блокировки ферментных систем. При полном нарушении процессов гидроксилирования наступает смерть. Частичная блокировка вызывает гиперпродукцию 17 -гидроксипрогестерона, прегнантриола, стероидов с андрогенными свойствами, что вызывает вирилизацию и нарушение водно-солевого баланса. |
Как выявить адреногенитальный синдром у детей
Диагностика расстройства основывается на фенотипических, анамнестических данных. Во время общего осмотра оценивают степень оволосения, состояние половых органов. Поздняя или неправильная диагностика врожденных форм адреногенитальных расстройств может привести к гибели ребенка в первые дни жизни. Проведение неонатального скрининга позволяет своевременно назначить адекватную заместительную терапию гормонами, правильно определить половую принадлежность младенца с синдромом.
Патогенетическим маркером расстройства служит определение 17-гидроксипрогестерона в образце крови, взятой на карту Гатри. Диагностическое значение имеют значения, превышающие 400 нг/100мл. Пренатальный скрининг адреногенитального синдрома проводится путем определения содержания 17-гидроксипрогестерона в амниотической жидкости. Возможность применения молекулярно-генетического анализа ограничена ввиду того, что существует более 10 мутаций, вызывающих блокировку синтеза 21-гидроксилазы. Из инструментальных методов диагностики применяются:
- рентгенография лучезапястных суставов – определяет имеющиеся деформации и устанавливает так называемый «костный» возраст больных с адреногенитальными расстройствами;
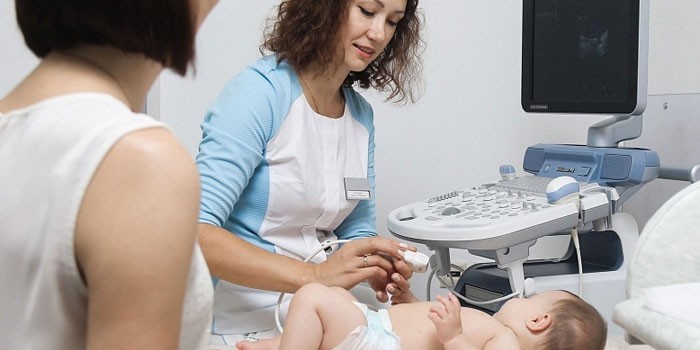
- УЗИ органов малого таза – выявляет патологии матки, яичников;
- томографические исследование надпочечников – применяются с целью выявления опухолевых процессов.
Методы лечения
Заместительная гормональная терапия – основной способ коррекции наследуемой дисфункции надпочечников. При ситуации, если у больной со скрытой формой адреногенитального синдрома нет репродуктивных планов, менструации ритмичны, а кожные проявления имеют минимальный характер, кортикостероиды не применяют. В остальных случаях выбор тактики лечения зависит от формы расстройства, жалоб пациентки. Как правило, глюкокортикостероиды дополняются другими лекарственными препаратами и хирургическими методами, подобранными согласно конкретной терапевтической цели.
Восстановление функции деторождения
При наличии репродуктивных планов женщине назначается гормональная терапия. Прием глюкокортикостероидов осуществляется строго под врачебным контролем. Гормональное лечение продолжается до полного восстановления менструального цикла и наступления беременности. Иногда дополнительно назначаются стимуляторы овуляции. Во избежание выкидыша прием кортикостероидов ведется до 13 недели гестационного срока. В первом триместре женщинам с адреногенитальными отклонениями рекомендованы эстрогены. Во втором и третьем – аналоги прогестерона без андрогенного эффекта.
Коррекция вирилизации
В случае если пациентка не планирует деторождение, но при этом желает избавиться от гирсутизма, угрей, нормализовать менструальный цикл, целесообразно применять препараты с антиандрогенным действием, оральные контрацептивы с гестагенами последнего поколения. Терапевтический эффект достигается через 3–6 месяцев с момента начала лечения синдрома. По окончании курса симптомы адреногенитального расстройства возвращаются.
Лечение ложного гермафродитизма
Пациенткам с такими проявлениями синдрома назначается гормонотерапия. Затем проводится хирургическая коррекция формы половых органов – вскрытие урогенитального синуса (интроитопластика), клитеротомия. При сольтеряющем адреногенитальном синдроме помимо глюкокортикостероидов под контролем активности ренина применяются минералокортикоиды с увеличением дозировок в случае возникновения интеркуррентных заболеваний.









Сообщить об опечатке
Текст, который будет отправлен нашим редакторам: